AI in Medtech: From Real-World Evidence to Content Generation
Originally published in: meditronic-journal (German edition), issue 5/2025
Co-authored by Jochen Tham, head of digital customer experience, ZEISS Medical Technology and Pearl Vyas, director, commercial content strategy, Veeva MedTech
Artificial intelligence (AI) advancements in the medtech industry have the potential to improve healthcare and patient outcomes by expanding the boundaries of new diagnostic capabilities, optimizing treatment pathways, and creating operational efficiencies.
However, this wave of innovation is met with significant uncertainty, as the future of AI in medtech is being actively shaped by a complex and evolving regulatory landscape.
With new stringent regulations like the EU AI Act set to take full effect for high-risk devices in 2027, many medtech companies find themselves at a crossroads, unsure how to use AI to improve their processes while remaining compliant.
The challenge is both technological and strategic. “Certain things can be sped up enormously by applying AI to them, but it’s not an end-to-end solution,” says Dr. Jochen Tham, head of digital customer experience at ZEISS Medical Technology.
Technologically, decisions about data inputs and how AI interacts with other tools in a company’s tech stack have cascading effects on costs and project timelines and can uncover deeper structural challenges within the organization.
Strategically, integrating AI requires a fundamental shift in thinking. “The way AI will do things is different from the way you have learned in the past,” Dr. Tham emphasizes.
This difference necessitates a significant cultural shift, requiring organizations to re-evaluate established roles, responsibilities, and workflows to fully harness the benefits of AI.
Decoding the impact of the AI Act on medtech
The EU AI Act introduces a substantial new element to what Erik Vollebregt, a partner at medtech-focused law firm Axon, aptly calls ‘regulatory lasagna.’ “In medtech, we are used to working with a degree of regulatory lasagna when regulations like MDR and the IVDR converge with other horizontal legislation like the Machinery Regulation or EMC Directive.
But the AI Act adds another layer of complexity,” explains Vollebregt. This complexity means medtech companies will likely have to undergo multiple conformity assessments and obtain parallel CE markings, a process that threatens to slow down an already lengthy and costly path to market.
This regulatory burden is not just a theoretical concern. It has prompted industry-wide calls for simplification to prevent stifling innovation. The industry association MedTech Europe has warned that the immense pressure of the new regulations disproportionately affects small and medium-sized enterprises (SMEs).
MedTech Europe reports that over 90% of the region’s medical technology manufacturers are SMEs, creating a critical issue as these companies are often the primary drivers of groundbreaking innovation.
Navigating these complex, multi-layered regulatory frameworks risks diverting resources away from R&D and slowing the introduction of novel technologies to market.
This is now compounded by the phased adoption of the AI Act. “Right now, we’re still dealing with the MDR and the IVDR being phased in. When you add in the AI Act, it becomes extremely challenging to develop and launch a product that will comply with the regulations as they continue to evolve,” Vollebregt notes.
Beyond product development and conformity assessment, this regulatory layering directly affects medtech companies’ commercialization efforts, particularly related to claims and promotional content. The issue isn’t that these regulations are inherently unreasonable, but rather that they introduce additional layers of complexity and ambiguity into commercial operations.
First, MDR Article 7 already establishes a high bar, strictly prohibiting any form of misleading advertising. Second, the AI Act introduces new, stringent obligations for transparency whenever AI is used to generate or personalize content.
This creates a high-stakes compliance environment where every piece of promotional copy, every AI-driven physician engagement, and every automated translation must simultaneously meet the MDR’s standard for accuracy and the AI Act’s standard for transparency.
This new reality necessitates deep expertise within commercial teams to evaluate marketing practices with a careful eye. The core challenge is that regulatory interpretation can be a subjective experience, often influenced by the specific knowledge and risk tolerance of individual reviewers. In the face of this ambiguity, commercial and regulatory teams take a cautious approach that can directly impact commercialization efforts already strained by significant operational friction.
Current industry data reveals a system under pressure. The review and approval process for promotional content is already notoriously slow – four weeks or longer for 35% of companies and typically undergoing three to five rounds of reviews before final approval. A lack of foundational infrastructure is a primary cause for delay, as nearly 70% of medtech organizations use manual or fragmented systems for managing claims and evidence.
The introduction of AI-specific regulations will likely act as a multiplier on these existing challenges, as the multidisciplinary teams responsible for approvals (medical/clinical, legal, regulatory) devote resources to comply with new standards for transparency and accuracy.
Real-world evidence in the context of EU MDR and the AI Act
MDR and AI Act requirements do converge in key areas, and it is here that a powerful solution emerges. James Dewar, co-founder and CEO of Scarlet, the only EU notified body specializing in software and AI, identifies real-world evidence (RWE) as an important tool for meeting these combined demands.
“RWE is derived from real-world data, which monitors how your device is performing in the real world,” says Dewar. “Once you analyze and annotate the real-world data, it turns into RWE.” RWE can satisfy multiple AI Act requirements.
For example, medtech companies can proactively plan and execute an RWE study as part of their conformity assessment to demonstrate that their device remains safe and effective over the long-term, directly addressing a core regulatory concern. For post-market surveillance, RWE provides a unified mechanism to satisfy the requirements of both regulations.
RWE as a commercial differentiator
For commercial teams, RWE provides a defensible and auditable foundation for all promotional narratives.
By systematically linking marketing claims within content management to annotated real-world datasets that support them, companies can create a transparent chain of evidence.
This practice not only demonstrates clinical relevance and ensures compliance with regulatory standards but also builds trust with health care professionals who increasingly seek evidence-oriented medical communications.
Furthermore, the AI Act mandates an assessment of fundamental rights, requiring companies to prove they have taken measures to mitigate risks an AI system may post to its users. RWE can be a specific instrument here.
“By monitoring your device in a real-world setting, you’ll be able to get data on the specific populations and subgroups that are actually using your device,” Dewar explains. “This will help you understand risks, like bias, in a much more granular way”.
As AI-driven personalization becomes more common in marketing, RWE acts as an essential safeguard against bias, ensuring that claims are accurate for, and resonate with, specific patient populations.
A strong RWE program can help “speed up certification lead times and AI feedback loops when retraining models based on new data or issue detection”. As Dewar states, “Being able to rapidly detect, collect, and analyze data from the real world is the key to building great AI products.”
Using AI to improve medtech content generation
Although the promise of AI is significant, its practical implementation is another challenge. A recent MIT study on generative AI adoption estimated that 95% of organizations are getting zero measurable return despite $30–40 billion in enterprise investment.
The report concludes that this stark divide between the few who are succeeding and the vast majority who are stuck is not driven by model quality or regulation, but is almost entirely determined by the strategic approach taken.
This finding serves as a reality check for the medtech industry. It suggests that the common perception that AI can be “sprinkled” on top of existing processes is fundamentally flawed. This is because AI operates on the principle of “garbage in, garbage out”. According to multiple industry reports, data scientists spend up to 80% of their time simply cleaning and preparing data before it can be used for analysis or AI modeling.
For example, in the case of medtech marketing, attempting to deploy generative AI for content generation (currently the most common use case in marketing) on top of fragmented, manual, or siloed content and claims management systems is a recipe for amplifying chaos and compliance risk, not solving it.
The successful 5% of organizations are not starting with AI; they are starting with their data and process foundation.
A prime example is ZEISS Medical Technology, which faced a complex and inefficient content process with end-to-end lead times ranging from weeks to four months. Their solution was not to jump directly to AI.
Instead, they undertook a foundational transformation, partnering with Veeva MedTech to first establish a centralized, end-to-end global content and claims management process. By implementing Veeva PromoMats as a single source of truth, they focused on harmonizing workflows, establishing clear governance, and creating a connection between claims and their substantiating evidence.
In essence, they solved the “garbage in” problem first, ensuring that any future AI system would draw from clean, compliant, and fully traceable information.
Within just three months, ZEISS Medical Technology achieved:
- A 50% faster time-to-market for content
- An 18-30% reduction in review and approval times
- 100% traceability of all promotional assets
Only after building this compliant, structured, and efficient content engine did ZEISS Medical Technology begin developing a generative AI tool for compliant text creation.
Their future architecture envisions AI as a powerful enabling layer that draws from the single source of truth, pulling approved claims, brand templates, and other verified content to automate and personalize campaigns.
The ZEISS Medical Technology journey shows that a successful AI implementation is the final step in a strategic journey, not the first.
It requires a solid foundation of data, process, and governance to deliver real value. As regulatory bodies continue to release guidance on AI/ML in healthcare, this ability to demonstrate data integrity and traceability will likely become the defining factor not only for ROI, but for market approval itself.
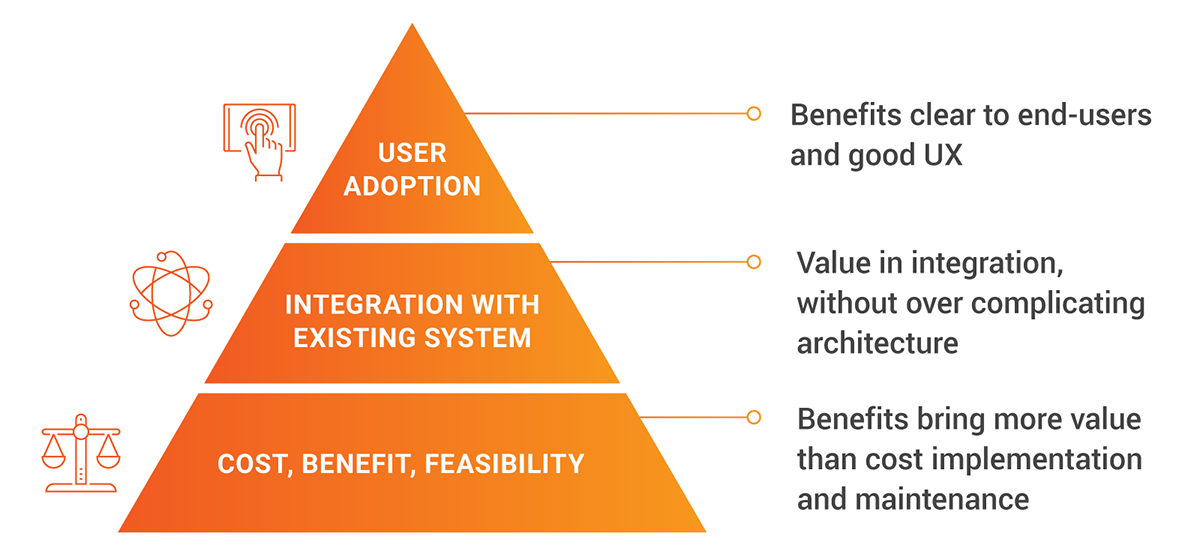
Winning in the new regulatory landscape
The convergence of advanced AI technologies with a multi-layered regulatory environment marks a pivotal moment for the medtech industry.
The central challenge is primarily the ambiguity and complexity of the new rules, as medtech commercial operations are already burdened by inefficient, manual, and fragmented processes.
This dynamic creates a significant risk of increased compliance violations, delayed market access, and a stifled innovation, especially for SMEs.
The current challenges are encouraging companies to create a new blueprint for future success.As the ZEISS Medical Technology case study demonstrates, the prerequisite for leveraging advanced technology like generative AI is not the sophistication of the algorithm but the strength of the operational foundation.
Companies that succeed will be those that address their foundational data and process gaps first, transforming their content and claims management from a siloed, manual function into a centralized, evidence-driven strategic asset.
This approach ensures that AI is used to amplify compliance and efficiency rather than chaos and risk.
Medtech leaders seeking to navigate this new landscape and build a competitive advantage can:
Elevate RWE to a core commercial asset.Transform RWE from a compliance cost into a competitive advantage. By making it the defensible and auditable foundation for every promotional narrative, companies can proactively satisfy both MDR and AI Act requirements while building crucial trust with healthcare providers.
Establish a centralized content foundation. Manual processes and siloed content are untenable in the face of layered regulations. A unified digital platform that serves as a single source of truth is the prerequisite for achieving compliance at scale and establishing a viable foundation for deploying AI effectively.
Deploy AI to amplify, not to originate. Treat AI as a powerful accelerator and not an end-to-end fix. The goal is to scale the personalization and localization of marketing assets that are derived from an existing library of approved, RWE-backed content. This avoids the critical data quality risk and ensures AI amplifies compliant messaging, not chaos.
Ultimately, the medtech companies that succeed will transform this regulatory challenge into an opportunity. By mastering the synergy between RWE, a compliant platform foundation, and strategic AI deployment, companies can move towards ensuring compliance while forging a new standard of trustworthy, evidence-based communication that builds a lasting advantage.
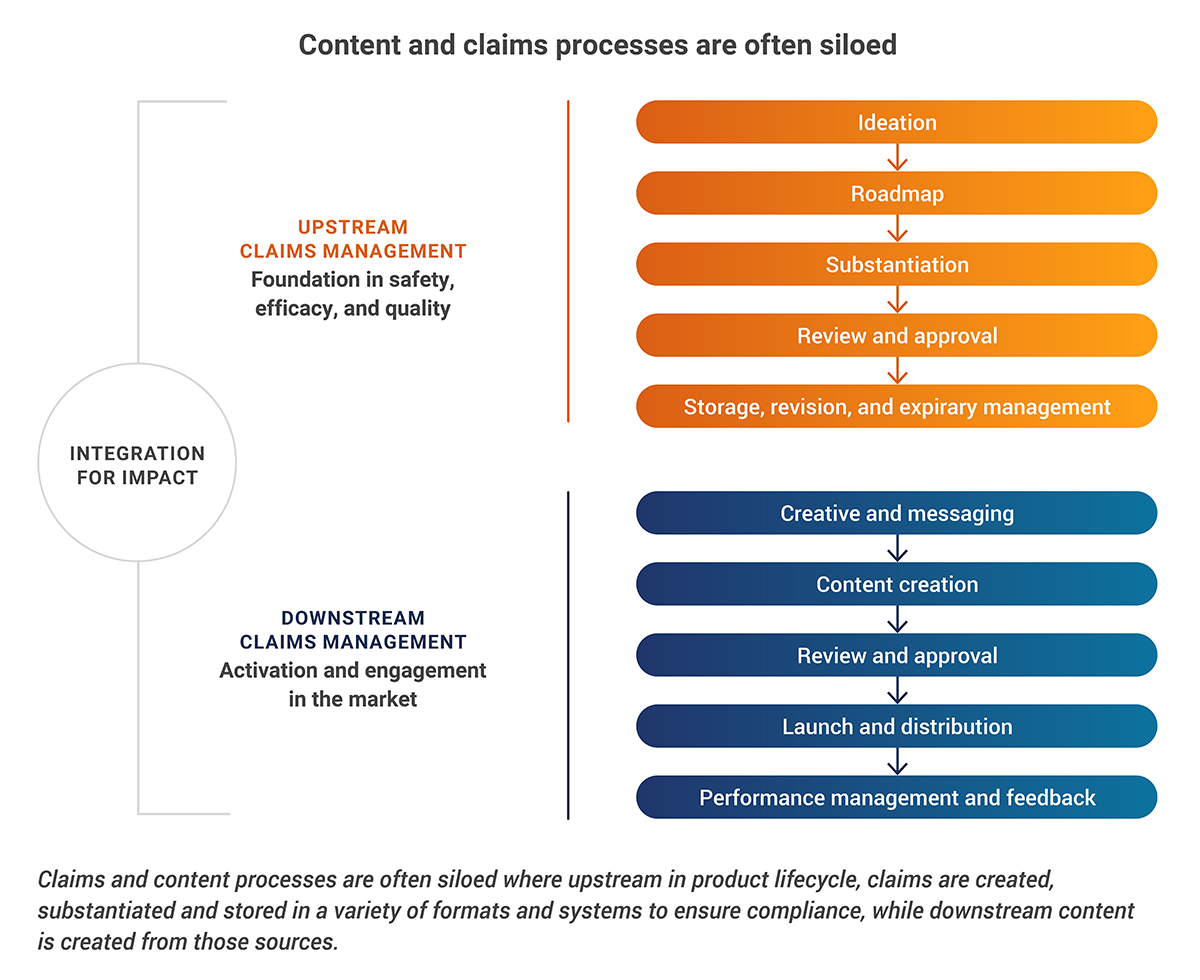
To learn more about how ZEISS is approaching AI initiatives, watch their full Summit session on Veeva Connect.