Ten Elements for a Streamlined Digital Regulatory Future
Abstract
Medical device and IVD regulatory systems rely on legacy, document-centric regulatory submissions. In practice, this means large volumes of information are recreated, reformatted, checked manually, and resubmitted in different ways across jurisdictions and review bodies. This results in too much time spent managing submission lifecycles, rather than assessing critical priorities: product safety, performance, and timely patient access.
With this in mind, the ABHI and Veeva MedTech co-hosted a regulatory executive roundtable, convening more than 20 senior leaders from medical device and IVD manufacturers, consultants, approved/notified bodies, and trade associations to define the essential elements of a modernized, proactive digital regulatory framework designed to streamline global conformity assessments.
This paper details a practical case for a unified digital model that is built on structured, reusable data instead of static document packages and is both open standards-based and solution-agnostic.
Introduction
Across the medical device and IVD sector, a persistent challenge has been the continued reliance on document-based submissions that are resource-intensive, difficult to maintain, and poorly suited to the scale and complexity of modern regulation. Reflecting the collective experience behind this paper, spanning more than 300 years in global submissions, the issue is not the absence of data, but the fact that critical regulatory information is too often locked within static, narrative-driven documents. This creates duplication for manufacturers, avoidable inefficiency for regulators and notified bodies, and a process that can divert attention from the central purpose of regulation: ensuring safety, performance, and timely patient access to innovation.A more modern approach is now needed, based on structured, interoperable data that can support clearer submissions, more consistent review, and greater efficiency across the product lifecycle.
To maintain global competitiveness and accelerate patient access to innovative products, medtech manufacturers need to move beyond static PDFs that lock data in an unusable state to a simplified, system-agnostic digital framework. This evolution is fundamental for a transparent, compliance-by-design model.
Replacing manual, document-centric workflows with an integrated, data-first system shifts the focus from navigating administrative hurdles to proactively ensuring the safety and performance of life-saving devices and diagnostics. Embracing these opportunities allows the medtech industry to resolve structural inefficiencies and accelerate patient access through a standardized electronic submission framework.
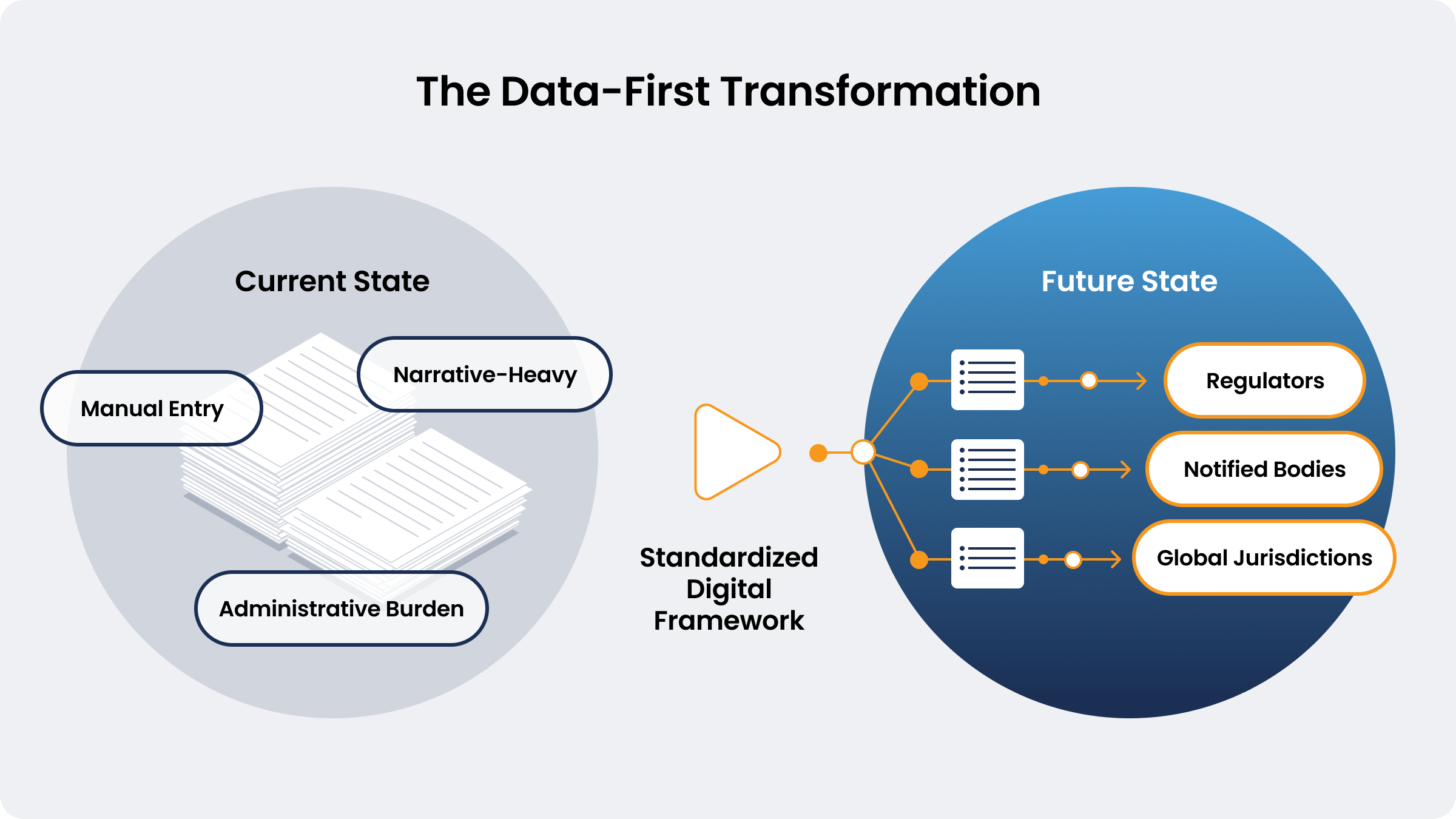
Traditional submission formats lack the structure needed for automated review and data or document exchange, leading to:
- Manual Data Entry: Teams spend significant time and resources duplicating, reformatting, and rectifying data within and across submissions. The European Commission estimates that over 30% of professional staff time in conformity assessments is dedicated to administrative tasks instead of core clinical, technical, and compliance reviews.
- Process Complexity: Regulatory frameworks such as the EU MDR and IVDR require extensive and at times duplicative documentation. This is often the result of duplication, where the same information is utilized in different contexts within the regulation. Furthermore, one of the main reasons for rejection of applications for conformity assessment is submission incompleteness.
Value of a Data-First Digital Framework
A standardized, system-agnostic digital framework would deliver clear benefits across the healthcare ecosystem, such as:
- Accelerated Market Access: A digital framework would shorten approval timelines and reduce turnaround time for post-market changes, allowing innovative products to reach patients faster. A standardized format can facilitate reviews and allow for immediate and complete identification of areas of change when submissions are updated.
- Enhanced Regulatory Capacity: Reducing manual burden and bureaucratic noise can free up capacity within both manufacturers and regulatory bodies, allowing teams to focus on identifying and addressing actual safety issues rather than administrative documentation inconsistencies. Further, the standardization of data format can enable additional efficiency gains through the use of AI. This is especially important for SMEs who have limited regulatory capacity but form 90% of the industry.
- Operational Efficiency: A standardized framework can provide a clear return on investment by lowering the cost of compliance, both for SMEs and large manufacturers alike, helping prevent good ideas from running out of funding or reaching patients while waiting for certification decision or approval.
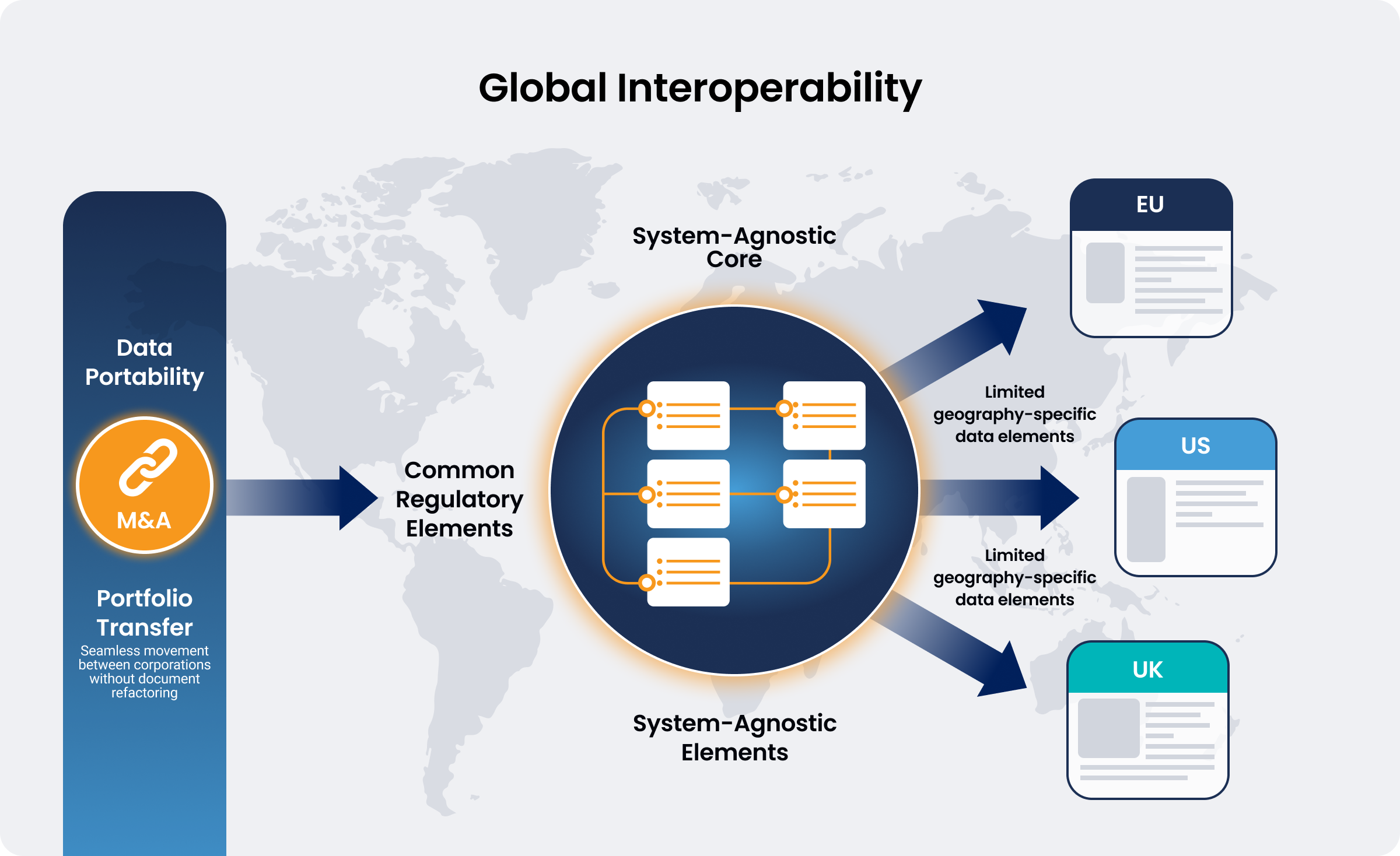
- Global Reliance and Scalability: A unified data foundation can enable frictionless data exchange across health authorities. If core regulatory elements are globally harmonized, a single structured dataset can support multiple geographies, requiring only minimal, region-specific data variants to scale globally.
- Data Portability: A standardized framework for data portability can support M&A activities and portfolio transfers. Technical documentation would no longer require manual refactoring to align with an acquiring company’s quality management system (QMS). Similarly, standardization can streamline transition between conformity assessment bodies.
- Increased Transparency: Well structured and traceable technical documentations can facilitate assessments/audits and enable easier maintenance through the product’s lifecycle.
Ten Elements to Streamline
This position paper outlines the ten essential elements required to build a streamlined regulatory future, centered on simplification and transparency. By moving from a reactive cycle of administrative correction to a proactive model of data-driven compliance, we can ensure a system framework that serves all stakeholders equally.
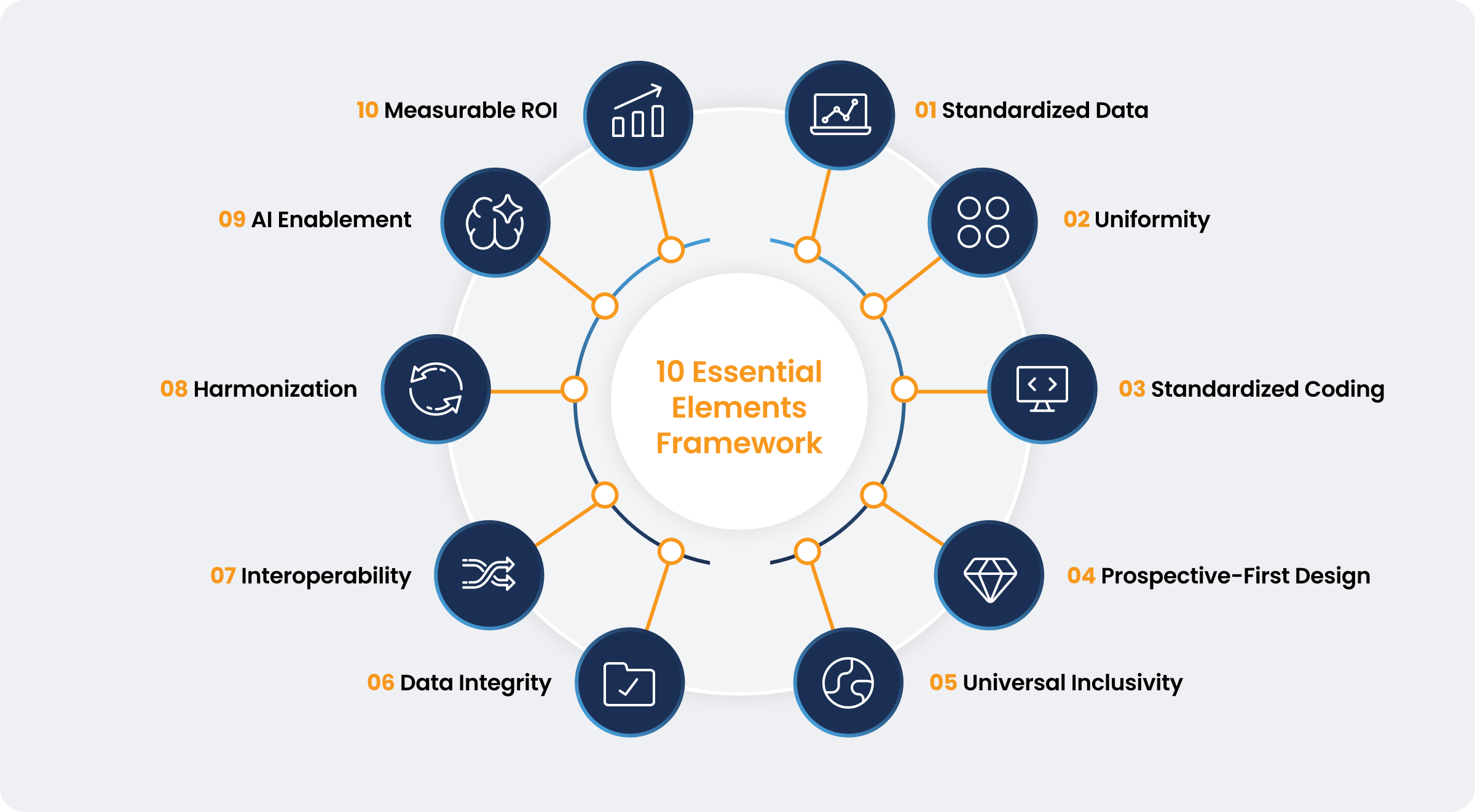
1. Standardized, Data-First Framework
The system must transition from narrative-heavy, unstructured documents to a standardized product-level dataset. This simplifies the submission process by reducing subjective interpretation, eliminating variation and error where elements are reused, and allowing for scalable and consistent interrogation by regulators or approved/notified bodies.
2. Uniformity Across Stakeholders and Lifecycle Stages
A single source of truth provides consistent information to manufacturers, regulators, and approved/notified bodies. This uniform submission layer allows all parties to leverage conformity assessment outcomes from other jurisdictions without full reassessment of underlying evidence, reducing parallel or sequential assessments of substantively similar technical documentation. Clear data definitions also ensure consistent information use across different lifecycle stages within the same organization or jurisdiction.
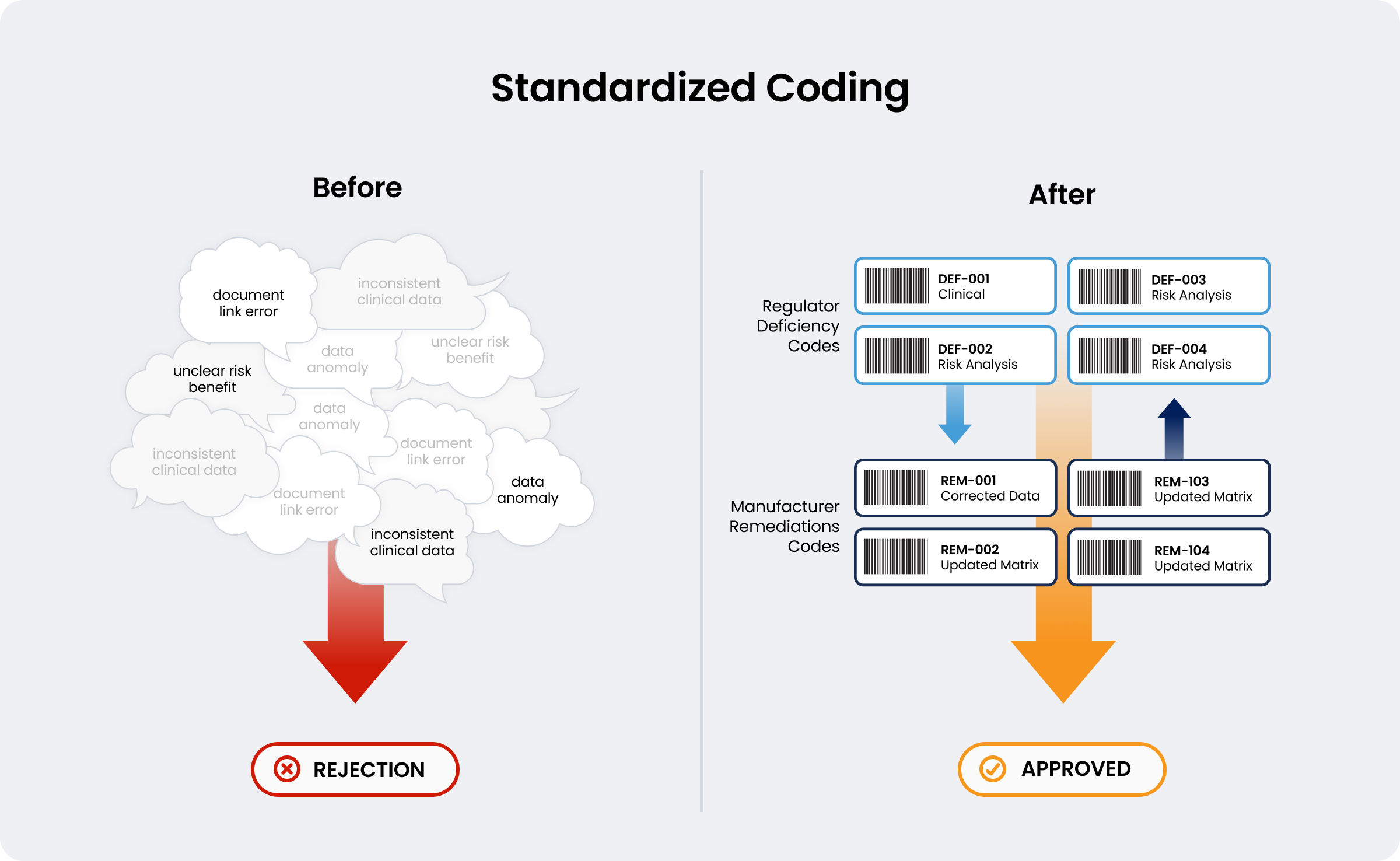
3. Standardized Coding for Deficiencies, Findings, and Remediations
Utilizing a harmonized global coding system provides unprecedented transparency into submission queries and rejections. Similarly, there should be reciprocal coding from the manufacturer denoting what has been done to address the issues raised. This dual-coding mechanism allows stakeholders to address systemic bottlenecks proactively before filing. Standardizing these data insights drives continuous improvement of global conformity assessments.
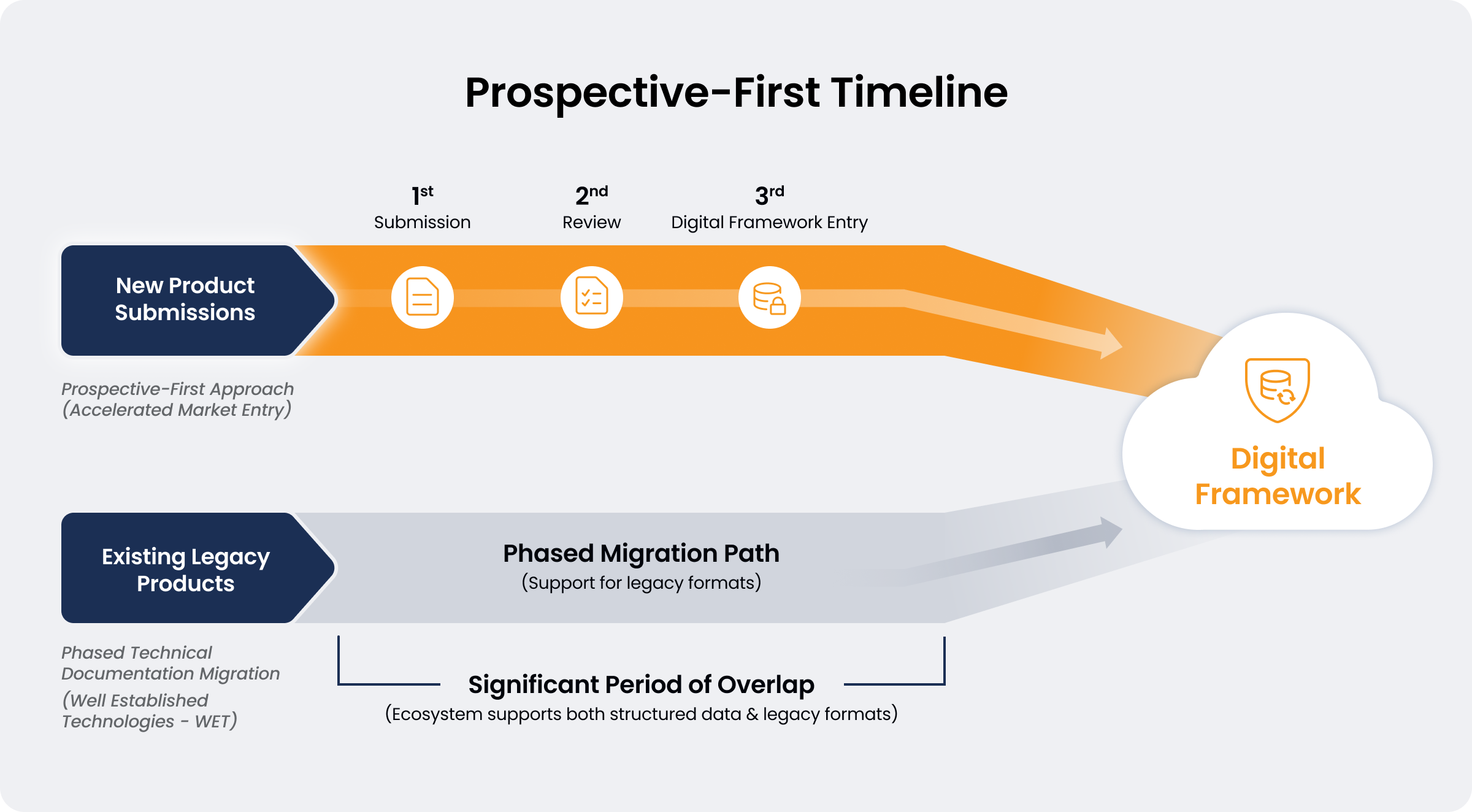
4. Prospective-First Design
The digital framework should prioritize a prospective-first approach for new product submissions to accelerate market entry for innovation. Migrating existing legacy technical documentation into the new framework requires a phased approach, meaning the ecosystem must support both legacy formats and structured data during a significant transition period. This approach avoids disproportionate resource implications of retrospective digitalization for legacy medical devices and IVDs, particularly for Well Established Technologies (WET).
5. Universal Inclusivity
The framework must be free to access and available for startups, SMEs, and large companies alike. This open accessibility supports inclusive regulatory participation and avoids imposing disproportionate short-term compliance costs on smaller market entrants.
6. Data Integrity and Lifecycle Governance
To ensure the reliability of regulatory outputs, the system enforces strict data integrity standards that shift the operational focus from reactive correction to proactive lifecycle management. This architecture prevents the submission of inconsistent or non-compliant datasets, ensuring that critical information, such as post-market surveillance data, is accurately captured and trended throughout the device lifecycle. Automated validation confirms that submission sections are complete, structurally sound, and audit-ready at every stage, as well as ensuring that a submission that meets the validation standards is automatically a complete submission.
7. System-Agnostic Portability and Interoperability
The framework must be built on open-source principles and global data interoperability to ensure vendor-agnostic portability, with due regard to confidentiality, controlled access, and cyber security. This capability is critical for portfolio transfers during corporate housekeeping, M&A activities, co-development arrangements, and moving between different jurisdictions. Such transfers should be able to be implemented without substantive reformatting of documentation, as well as enabling manufacturers to transfer their regulatory data across systems or organizations, as required.
8. Global Harmonization
The roadmap to success should be global, leveraging frameworks like the IMDRF Reliance Playbook. Implementation should follow a phased, scalable pilot approach to progressively expand standards over time. To achieve true global alignment, this digital infrastructure must remain independent of specific regulators or conformity assessment bodies.
9. Artificial Intelligence Enablement
Structured data provides the robust foundation required to successfully apply AI. This enables medtech manufacturers and regulatory body professionals to focus on value-added activities required for increasingly complex evaluations, such as assessing safety and effectiveness of medical devices and IVDs.
10. Measurable Return on Investment
The framework must drive efficiency through the simplification of the review process, reducing administrative duplication of assessment activity and shortening review timelines both in a single jurisdiction and across markets. This frees up capacity within manufacturers and regulators or approved/notified bodies alike, supporting high value innovation over administrative remediation.
Conclusion
As healthcare rapidly transforms, the medtech industry’s reliance on outdated, document-centric workflows creates a widening gap between regulatory practices and technological progress. By adopting these ten elements, medtech manufacturers, regulatory authorities, and approved/notified bodies can move beyond the burden of reactive administrative correction, support manufacturers of all sizes, and embrace a model of proactive compliance by design. The transition to a digital regulatory system is therefore a necessity for medtech. This evolution centers on simplification and a transparent, data-rich environment where expectations are clear and outcomes are predictable. By ensuring this framework remains entirely system-agnostic, this digital regulatory approach protects the portability of innovation and prevents the fragmentation of global markets. A harmonized digital foundation will not only accelerate patient access to life-saving technologies but also provide the necessary infrastructure for safe and effective AI integration.
Moving beyond pilot programs requires a commitment across the industry to implement a unified global roadmap creating operational efficiency for all. By uniting around this shared vision, the medtech ecosystem can transform regulatory compliance from a bureaucratic hurdle into a strategic accelerator-one that upholds the highest standards of product safety and performance while ensuring timely patient access to life-saving innovations.

CO-AUTHORS
| Bassil Akra AKRA TEAM GmbH |
James Dewar Scarlet |
Sarah Haake-Schäfer CZ Vision |
Crystal Allard Veeva Systems |
| Cait Gatt BD |
Diogo Geraldes Veeva MedTech |
Rita Hendricusdottir RegMetrics |
Rita Peeters J&J |
| Roland Back Abbott |
Megha Deviprasad Iyer Thermo Fisher Scientific |
Sinead Quaid Cook Group |
Deniz Bruce GMDN |
| Nina Goddard Intuitive Surgical Ltd |
Steve Lee ABHI |
Amra Racic Veeva MedTech |
Anastasia Stergiadi J&J |
| Tom Patten GMED |
Benjamin Rochette Coloplast |
Darren Thain Smith & Nephew |
James Shearn STERIS Corporation |
| Erik Vollebregt Axon Lawyers |
Sue Spencer Compliance Connextions |
Erin Wigglesworth Danaher Corporation |