Getting Ahead of EUDAMED Implementation Timelines
Implications for Enterprise Regulatory Information Management
By Diogo Geraldes, Director, EU Regulatory Strategy, Veeva MedTech
The European Commission recently confirmed the operational status of key EUDAMED modules, establishing fixed regulatory compliance deadlines. Effective May 28, 2026, organizations are required to transition from national processes to centralized EUDAMED registration for actors, devices, certificates, and related systems/procedures. All devices placed on the EU market after this date will require successful EUDAMED registration prior to commercial release. A defined transition period applies until November 28, 2026 for the migration of legacy device records into EUDAMED. These timelines establish statutory compliance milestones with direct regulatory, operational, and commercial impact.
Operational challenges in achieving compliance
For large medical technology enterprises, these requirements create substantial cross-functional operational, system, and governance demands. Managing large SKU portfolios and complex device families requires treating the EUDAMED transition as a comprehensive enterprise data management initiative. Regulatory data is often decentralized across disparate internal platforms, including ERP, PLM, and QMS, as well as disconnected spreadsheets. Establishing validated, secure machine-to-machine interfaces for high-volume regulatory submissions imposes distinct technical and operational burdens. Treating UDI submissions as isolated IT activities rather than integrated enterprise regulatory processes may increase long-term compliance, audit, and business continuity risks.

Many organizations rely on standalone UDI tools that operate independently of core regulatory and enterprise systems. This fragmented model often fails to address broader data lifecycle requirements. UDI functions as a regulated enterprise data asset that spans the full device lifecycle, from development through post-market surveillance. UDI data typically resides across multiple regulated and operational enterprise systems, including:
- QMS: For complaints, nonconformances, and CAPAS.
- Supply chain: For product traceability, recalls, and shortage management.
- Clinical: For implant cards and post-market clinical follow-up.
- Regulatory: For certificates, declarations of conformity, and market authorization.
Siloed UDI data may increase the risk of integration failures, regulatory inconsistencies, adverse audit findings, and delayed market access. This disconnected approach results in manual data transfer, increased validation burdens, and higher error rates.
Integrated UDI and regulatory information management strategies
Integrating UDI management directly into enterprise regulatory information management (RIM) platforms offers a more robust alternative. In this model, UDI serves as a central component of the regulatory strategy rather than a supplementary function. Integrated regulatory platforms enable organizations to gather and analyze data across quality, clinical, and regulatory teams, consolidating regulated product data into a governed, auditable system of record.
A unified approach may incorporate the following elements:
- Centralized master data management of core UDI attributes: Capture common UDI attributes once (brand name, clinical size, storage) to be used for all markets.
- Registration management: Link UDI master data directly to regulatory submissions, change control, and market authorization activities.
- Validation and submission: Generate UDI data in the Health Authority format, validate it against regulatory and business rules, and submit it to EUDAMED through controlled, auditable system processes.
Global regulatory integration considerations
Regulatory and UDI compliance requirements extend beyond the European Union and will continue to evolve across global markets.
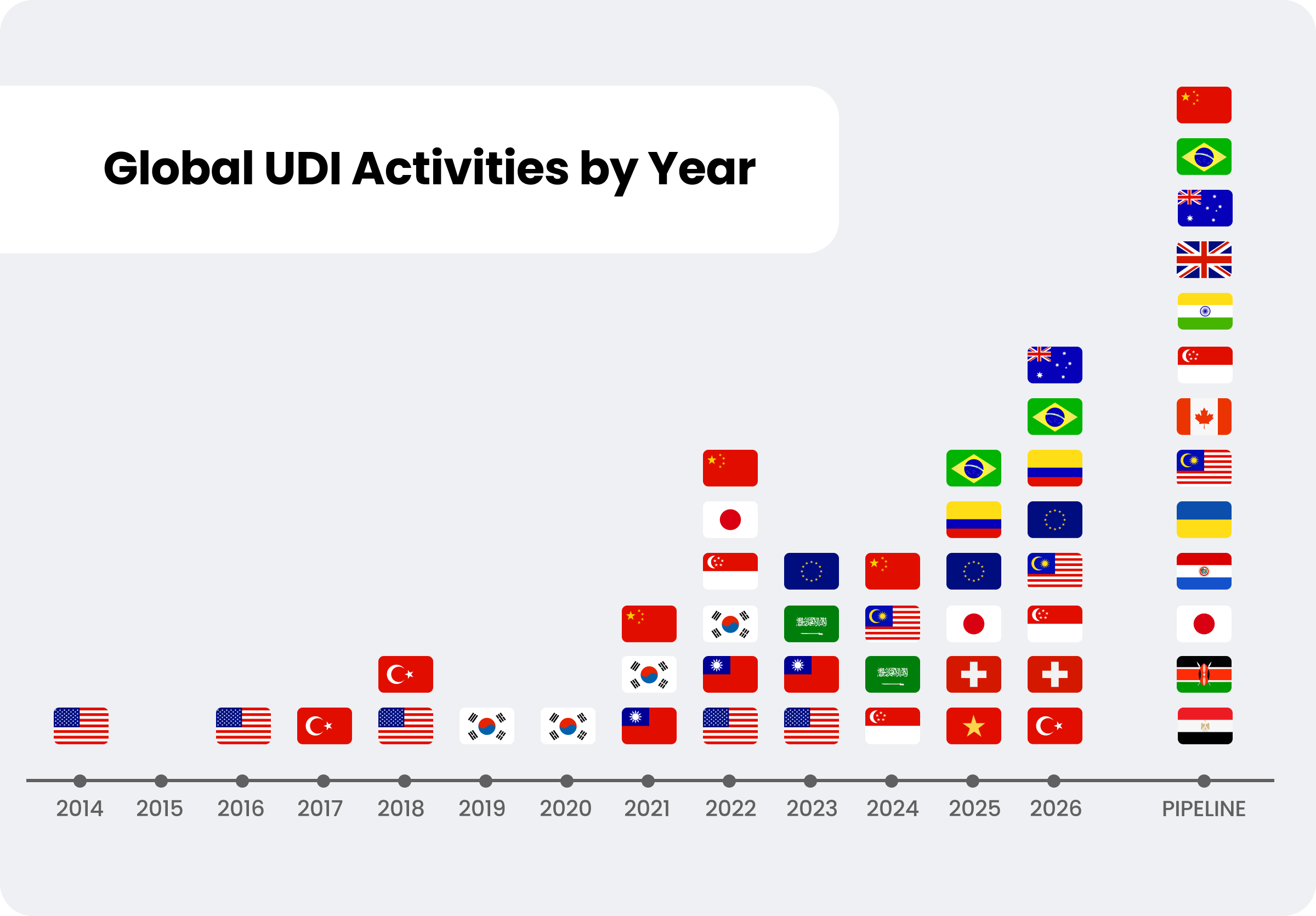
Veeva RIM supports EUDAMED data preparation, regulatory event management, and machine-to-machine submission readiness. By continually expanding UDI capabilities, Veeva Medtech is committed to supporting multi-authority submission, controlled data governance, and global regulatory scalability. We also regularly gather feedback from a customer working group to help define our UDI roadmap, market prioritization, and general UDI export features.

Strategic and operational considerations
The confirmation of EUDAMED timelines underscores the importance of transitioning from manual regulatory processes to centralized, system-driven digital compliance approaches. Organizations may benefit from platforms that integrate UDI and regulatory data throughout the product lifecycle, from design control through post-market vigilance, ensuring alignment across multiple functions. Organizations should assess whether their regulatory, data, and system architectures are sufficiently centralized and governed to support EUDAMED compliance by May 2026 and leverage this infrastructure as the foundation to then expand compliance for the vigilance module.
—Next: Learn how to tackle complex medtech regulations using the resources at your disposal.