製薬会社が医薬品開発のプラットフォーム導入を検討すべき理由
医薬品開発効率化におけるIT課題
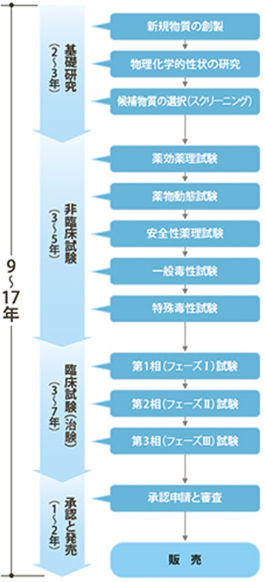
医薬品開発は、基礎研究から始まり、非臨床試験、臨床試験(治験)、承認申請・審査、上市という流れで進む。新有効成分含有医薬品が医療機関を通じて患者に届けられるまでに、9~17年ほどかかるのが一般的だ。膨大な時間と費用をかけて1つの新医薬品が世に出るわけだが、その過程で幾つもの遵守事項があり、それらは法制化されている。ヒトでの有効性及び安全性を調べる臨床試験(治験)に先立ち、動物や培養細胞等を用いて新規物資の有効性・安全性を研究する非臨床試験においては、試験施設(場所)の設備・機器、組織・職員、検査・手順・データ取得等が適切であり、データの質と信頼性を確保する目的で「Good Laboratory Practice」(優良試験所規範、略して「GLP」)が規定されている。同様に、臨床試験の実施では「GCP(Good Clinical Practice)」、製造管理・品質管理の基準として「GMP(Good Clinical Practice)」、品質試験では「GQP(Good Quality Practice)」が適用される(これらの規定は総称して「GxP」と呼ばれる)。医薬品開発は患者の健康と生命に関わるため、有効性、安全性、品質などを確認する各試験実施とそれら報告書作成の過程について、きめ細かく厳格な法制度の下に実施されるのだ。また、上市後においても、「GMP」「GQP」に加えて、医薬品安全性確保に関わる事項では「GVP(Good Vigilance Practice)」、製造販売後調査・試験の実施基準では「GPSP(Good Post-marketing Study Practice)」という規制がある。
製薬企業は、こうした医薬品開発における要求事項を満たしつつ業務効率化のため、業務対象に応じたシステム開発を大手システムインテグレーターへ委託し、独自システムを構築しているケースが多い。その結果、開発業務領域間でつぎはぎだらけのシステム群になってしまい、システム間のデータ連携ができず、一部工程を手作業で処理したり、再入力したりせざるをえない状況が生じているところもあるようだ。また、規制要件変更や技術進歩に伴う頻繁なシステム改修が必要となり、そのたびに手間と費用が発生してしまうという問題もある。
製薬企業に求められるグローバルスタンダードへの対応
新医薬品開発には膨大な開発コストがかかるため、自社で1から研究開発するよりも、ブロックバスター(従来の治療体系を覆す薬効を持ち、他を圧倒するシェアや新市場開拓により莫大な利益を生み出す新薬)を持つ海外有望企業を買収したほうが手っ取り早いという考え方もある。大手製薬会社の中にはM&Aを中核戦略に据えてグローバル展開しているところもあるが、すべての製薬会社が同様にグローバル進出できるわけではない。一方、自社の医薬品開発プロセス全体をグローバルで標準化・効率化させ、開発コスト低減かつ開発期間短縮に注力することは、自社の状況に即して実践可能である。
通常、臨床試験後の承認審査期間だけでも1~2年かかるが、システムによる効率化によって、仮に1カ月早く承認申請ができたとしよう。この場合、ブロックバスターの医薬品であれば約30億円もの売上前倒し効果が期待できるというデータもある。
国内の製薬会社が欧米の大手製薬会社に遅れることなく対抗するには、もはや新たな視点によるシステム投資は避けることができない。欧米の大手製薬会社は、分断化されたシステム群から統合化されたプラットフォームへの移行を完了あるいは進行させているところである。もはや統合化されたプラットフォームはグローバルスタンダードとなっており、日本でも“待ったなし”で移行を進めるべき状況となっている。
最適解は一元管理可能なプラットフォームへの移行
製薬会社の医薬品開発向けのシステムプラットフォームはすでに複数のIT企業から提案されているが、何を基準に選択すればよいのだろうか。ポイントは、医薬品開発プロセスの全業務領域を包括的に一元管理できるかどうかである。同時に、各種規制に対応する機能要件を満たすシステムであることを検証(バリデーション)できなければならない。
すなわち、試験過程とその取得データの信頼性保証の観点から、利用するシステムの要件を明確にし(要件の文書化)、システムが意図したとおりに(期待される効果の文書化)、 動作すること(検証結果の文書化)を保証するコンピュタシステムバリデーション(略してCSV) が求められる。教育訓練等による適切な運用と併せて、電子データの内包するリスク、データの真正性、見読性、保存性等に関するリスクへの対応がなされ、データ品質が一貫して満たされなければならない。
Veevaでは、医薬品開発向けの「Veeva Development Cloud(Vault Platform)」という一元化されたプラットフォームを提供しており、すでに国内外で豊富な実績がある。同製品の特徴は、1つの共通プラットフォームの上に、それぞれの業務アプリケーションが開発されるところにある。そのため、CSVの負担を最小限に抑えることができるのだ。各国の規制要件を満たしているため、治験開始時の準備や各種申請書類の作成など各試験に付随する時間を短縮でき、作業を迅速化できる。また、年に3回の定期アップデートにより、最新のトレンドやテクノロジーも随時取り入れられている。コンプライアンス対応等によるシステムの見直しを迫られることもないため、安心して利用できるのだ。
医薬品開発の世界も継続的にイノベーションを取り込んでおり、その変化はますます早まっている。今後、日本の製薬会社がグローバル市場で勝負するためには、運用面でもグローバルで標準化・一元化されたプラットフォームが強力な武器になることはまちがいない。