Resource Center
AI (Veeva AI)
View More AI (Veeva AI) Resources



Clinical Data
View More Clinical Data ResourcesLearn More
Revolutionizing Clinical Data

Watch Video
Platform Over Patchwork: How Consolidation Drives CRO Efficiency

Learn More
Survival of the Fastest: A Practical Framework for Risk-Based Data Management

Watch Demo
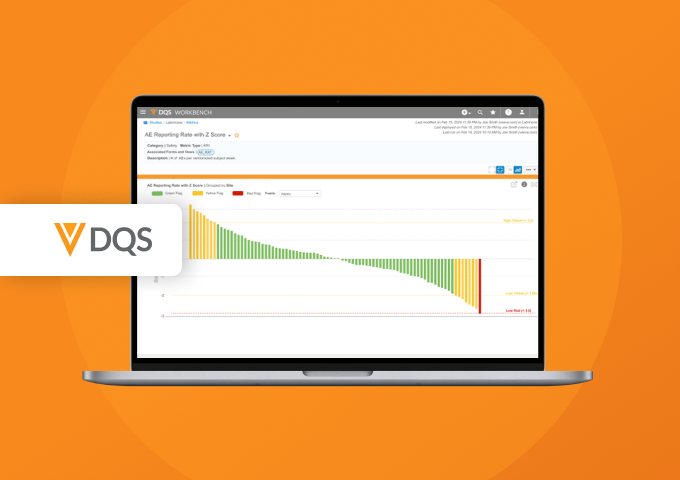
Veeva DQS Feature Highlights: Protocol Deviations and More
Clinical Operations
View More Clinical Operations Resources



Commercial
View More Commercial ResourcesWatch Video
Commercial Evidence™ in Vault CRM and Ostro
Read Report
Veeva Agentic Commercial Trends Report

Watch Video
Commercial Evidence Generated from the Field with the Agentic Call Report™

Watch Video
Commercial Evidence from Conversational AI for Patients and HCPs with Ostro

Learn More
Top Launch Challenge: Accelerating Field Activation

Watch Video
Advanced Targeting and Field Insights with Vault CRM

Learn More
2026 ASCO Insights Powered by Veeva Link Data

Watch Video
Bayer AG: AI-Driven Insights Through Connected Data and Software

Development Cloud
View More Development Cloud ResourcesWatch Demo
Submission Document Collection
Learn More
Discover Untapped Value with Strategic Efficiency Optimization in Drug Development

Watch Video
Falcon: Agentic Labor That Delivers Business Outcomes

Watch Video
Keynote Highlights: Enabling Agentic Biopharma™: AI Agents Working For You and Beside You

Medical
View More Medical Resources


Quality
View More Quality ResourcesRead Press Release
Veeva Introduces New EHS Application

Read Press Release
Veeva Quality Cloud Advances Operations for Kindeva

Learn More
Accelerate Lead Times with External Collaboration in Quality

Learn More
The Essential Ingredient for GxP Training Innovation

Regulatory
View More Regulatory Resources



Research Sites
View More Research Sites ResourcesLearn More
6 Strategies to Simplify Study Training for Sites

Read Press Release
Veeva Announces eSource Application to Eliminate Paper

Read Press Release
Veeva and SCRI Form Strategic Collaboration

Read Press Release
Veeva Announces Research Site CTMS

Safety
View More Safety Resources



Vault Platform
View More Vault Platform ResourcesWatch Video
Integrating Agentic AI Into Veeva Quality Cloud

Watch Video
Keynote Highlights: Vault AI: Productivity Built Into Every Vault Application
Read Press Release
Teva Pharmaceuticals Commits to Veeva Vault CRM

Learn More
Copying Vault Data Using Direct Data API and Migration Mode

Result Not Found
The query that you've entered is not found.